عمل فیزیولوژیک پروتئین طبیعی نامعلوم است. شکل بیماریزای پروتئین (PrPsc) با تغییر شکل فضایی و سه بعدی شکل طبیعی به وجود میآید. این تغییر شکل ممکن است به علت جهش در ژنهای کدکننده پروتئین طبیعی در بدن باشد یا به علت ورود پریون (یا پروتئین غیرطبیعی) از طریق عفونت به وجود آید.
پریونها بر خلاف تمام اشکال حیاتی دیگر برای تکثیر به وجود DNA یا RNA وابسته هستند، با وجود آنکه صرفا از پروتئین تشکیل شدهاند، میتوانند خود را بازسازی کنند.
برای شناخت پریونها باید به تاریخچه گروهی از بیماریهای تحلیلبرنده عصبی مرگبار به نام "آنسفالوپاتیهای اسفنجیشکل مسری" بپردازیم.
بیماریهایی مرگبار با علت نامعلوم
دست کم از ۲۰۰ سال پیش یک بیماری شگفت آور در گوسفندان به نام اسکرپی(Scrapie) شناخته شده بود که با زوال پیش رونده دستگاه عصبی مرکزی همراه بود.
این بیماری که مسری بودن آن بین گوسفندان از بیش از یکصد سال پیش توصیف شده است، گوسفندان ۲ تا ۵ ساله را گرفتار میکند و دوره پنهانی (فاصله زمانی بین آلودگی با عامل بیماری، و ظهور علائم) طولانی مدتی بین یک تا دو سال دارد.
اولین علائم بیماری تغییرات رفتاری گوسفند مثل ناآرامی و بی قراری است، سپس حیوان دچار کاهش وزن و ضعف می شود، در سر و گردن لرزش پیدا می کند و هماهنگی عضلانیاش را از دست میدهد، گوسفند مبتلا شروع به خاراندن بدنش با اشیا میکند و نام بیماری هم از همین رفتار حیوانات مبتلا گرفته شده است (مالیدن= scrape)
حیوانات مبتلا در طول ۵ تا ۶ ماه میمیرند، در کالبدشکافی حفرات متعددی در مغز جانوران مبتلا و اسفنجی شدن آنها مشهود است. احتمال داده میشد که استفاده از مرتع مشترک باعث انتقال بیماری بین حیوانات شود.
در همان حال در دهه ۱۹۲۰ دو عصب شناس آلمانی هانس گرهارد کروتزفلد و آلفونس ماریا جاکوب یک بیماری نادرکشنده تحلیل برنده مغزی را در انسان توصیف کردند.
این بیماری مردان و زنان را به نسبت مساوی در حدود ۶۰ سالگی گرفتار میکرد. بیماران دچار علائم روانی و رفتاری مبهمی میشدند که پس از هفتهها و ماهها به زوال عقل پیش رونده منتهی میشد و اغلب با حرکات غیرطبیعی بدن و اختلال دید همراه بود.
بیماری درمان پذیر نبود و در کمتر از یک سال از شروع علائم به مرگ منتهی می شد. کالبدشکافی این بیماران هم ایجاد حفرات در مغز و اسفنجی شدن آن را نشان می داد. علت بیماری نامعلوم بود.
رازی که گشوده شد
درادامه ماجرا به دهه ۱۹۵۰ می رسیم که کارلتون گایدشک متخصص اطفال و بیماریهای عفونی از دانشگاه هاروارد در "انستیتو پاستور تهران" به پژوهش مشغول بود. او در سال ۱۹۵۵ از طرف "موسسه پزشکی والتر و الیزا" در ملبورن استرالیا به عنوان پژوهشگر بازدیدکننده دعوت شد و از تهران به ملبورن رفت.
او ضمن کار در این موسسه با بیماری عجیبی آشنا شد که در قبیله fore درکوهستانهای شرقی پاپوای گینه نو شایع بود و بومیان آنرا کورو (kuru) به معنای "رعشه" مینامیدند.
اولین علائم بیماری با درد مفاصل و سردرد شروع میشد و به طور شاخص با از دست رفتن هماهنگی عضلات، لرزش و زوال عقل ادامه مییافت. بیماری پس از شروع علائم به طور مداوم پیشرفت میکرد و دو سال پس از شروع علائم مرگ بیمار را باعث میشد.
کالبدشکافی این بیماران هم اسفنجی شدن مغز را نشان میداد. سالها بود که پژوهشگران اعتقاد داشتند که این بیماری وراثتی است، هر چند الگوی بروز بیماری که اغلب زنان بزرگسال و کودکان از هر دو جنس را مبتلا میکرد و مردان بزرگسال به ندرتً به آن مبتلا میشدند، برای یک بیماری وراثتی غیرمعمول بود.
گایدشک با زندگی در میان این قبیله و مطالعه کردن زبان و فرهنگ آنها به کالبدشکافی افراد فوت شده به علت این بیماری پرداخت. او توانست ثابت کند که بیماری ماهیت عفونی و مسری دارد نه وراثتی.
او نمونههایی از بافت مغزی افراد فوت شده از کورو را به مغز شمپانزه ها تزریق کرد؛ میمون ها نهایتاً دچار بیماری مشابه انسان شدند و به علت آن مردند. همچنین او توانست الگوی غیرمعمول بروز بیماری را توضیح دهد.
بیماری به علت مراسم آدم خواری افراد فوت شده که جزیی از مراسم سوگواری بود بین انسان ها انتقال می یافت. از آنجایی که زنان و کودکان در این مراسم مغز متوفی را می خوردند و عامل عفونی ناشناس که در مغز قرار داشت، بیماری بیشتر آنها را مبتلا می کرد.
گایدشک در سال ۱۹۵۹ به ریاست آزمایشگاههای ویروس شناسی و عصب شناسی "موسسههای ملی بهداشت" (NIH) آمریکا رسید و به تحقیقات خود در زمینه این دسته بیماری ها که اکنون "آنسفالوپاتیهای اسفنجیشکل" نامیده می شوند، ادامه داد.
نکته عجیب در انتقال تجربی بیماری به حیوانات این بود که عامل عفونی اسرارآمیز عامل بیماری در مقابل تابش اشعه ماوراءبنفش (که اسیدهای نوکلئیک را متلاشی می کند) مقاومت میکرد و تنها با عواملی که پروتئین ها را تجزیه میکنند، غیر فعال میشد.
او معتقد بود که عامل این بیماریها ویروسی با عمل تدریجی و کند است که شاید توانایی نهفته ماندن در بدن بیمار برای مدت طولانی را دارد و برای همین است که این بیماری ها دوره پنهانی طولانی و سیری کند دارند.
این ویروسها را ویروسهای کند (Slow viruses) نامیدند و گایدشک که علاوه بر پژوهش در ویروس شناسی در حوزه های مختلفی مانند یادگیری و رفتار، رشد کودک، تکوین فرهنگهای بدوی، ژنتیک و ایمنی شناسی فعالیت کرده بود در سال ۱۹۷۶ جایزه نوبل پزشکی را دریافت کرد.
اما ماجرا به اینجا ختم نشد. پنج سال بعد در ۱۹۸۲ ادعای استانلی پروسنر استاد عصب شناسی و بیوشیمی دانشگاه کالیفرنیای سان فرانسیسکو قضیه را پیچیدهتر کرد.
پروسنر در زمانی که دوره دستیاری عصب شناسی را میگذراند مسئولیت بیماری را به عهده داشت که به علت "بیماری کروتزفلد - جاکوب" (CJD) فوت کرده بود. این امر باعث جلب علاقه او به این طبقه کمتر شناخته شده بیماریهای تحلیل برنده عصبی یعنی "آنسفالوپاتیهای اسفنجی شکل" شد.
او در سال ۱۹۷۴ آزمایشگاهی را برای تحقیق در مورد بیماری اسکرپی بر پا کرد و در سال ۱۹۸۲ ادعا کرد که عامل ایجادکننده بیماری را جدا کرده است. پروسنر مدعی شد که عامل بیماریزا که او آن را "پریون" نامید، با سایر عوامل بیماری زا مانند باکتری یا ویروس متفاوت است زیرا تنها از پروتئین تشکیل شده و فاقد ماده ژنتیکی DNA یا RNA است که همه اشکال حیاتی برای تکثیر به آن نیازمندند.
نظرات پروسنر با ناباوری عمومی جامعه علمی روبه رو شد، اما حوادث بعدی صحت نظریه پروسنر را ثابت کرد.
جنون گاوی ظهور میکند
در سال ۱۹۸۶ بیماری کشندهای مشابه اسکرپی در میان گاوها در انگلیس همهگیر شد. این بیماری را که مانند اسکرپی با اسفنجی شدن مغز همراه بود آنسفالوپاتی اسفنجی شکل گاوی (BSE) نامیدند.
این بیماری دارای دوره نهفتگی طولانی بین ۲ تا ۵ سال و علائم آن شامل تغییرات رفتاری مانند هیجان زدگی و بی قراری حیوان و از دست رفتن پیش رونده هماهنگی عضلانی و کارکرد حرکتی حیوان است.
در مراحل پیشرفته بیماری کاهش وزن و انقباضات ظریف عضلانی در گردن و تنه حیوان بروز می کند و حیوان به شیوه ای غیرطبیعی و اغراق شده گام برمی دارد و از گله جدا می شود. مرگ معمولاً در طول یک سال از شروع علائم رخ می دهد و درمانی هم وجود ندارد.
این بیماری پس از ظهور در سال ۱۹۸۶، در جنوب انگلیس همه گیری ایجاد کرد و مواردی از آن در اروپا و کانادا هم گزارش شد. در هنگام ظهور BSE در مورد احتمال انتقال آن به انسان نگرانی هایی ایجاد شده بود.
در ابتدای ۱۹۹۶ مواردی از ابتلا به بیماری کروتز فلد- جاکوب در افراد جوانتر از سن معمول شیوع بیماری یعنی ۵۰ سالگی دیده شد و بررسیها نشان دهنده گونه جدیدی از CJD بود.
پژوهش وجود عامل ژنتیکی در بیماری را رد کرد و نشان داد که انتقال در نتیجه در معرض قرار گیری بافت های گاوهای مبتلا به BSE از طریق خوراکی است.
به این ترتیب گزارش پروسنر در انگلیس مورد توجه ملی قرار گرفت و نشان داده شد عامل پروتئینی مسری عامل BSE با" پرشی میان گونهای" به انسان منتقل شده است. در مورد خود BSE هم ظاهراً پریون عامل بیماری اسکرپی در گوسفندان از طریق اضافه کردن مکمل های غذایی پروتئینی به دست آمده از گوسفند های آلوده به غذای گاو ها باعث انتقال بیماری به آنها شده بود.
پروسنر در سال ۱۹۹۷ به خاطر پژوهشهایش در این حوزه جایزه نوبل پزشکی یا فیزیولوژی را دریافت کرد.
پروسنر در ابتدا پیشنهاد کردکه حضور پریون یا پروتئین تغییر شکل یافته به نوعی که کاملاً شناخته نشده است باعث ایجاد واکنشی زنجیره ای می شود که PrP های طبیعی را تغییر شکل می دهد و ذرات عفونی جدیدی به وجود می آورد. پروسنر در مقالات بعدی خود مکانیسم دیگری را برای تکثیر پریون ها در مغز پیشنهاد کرد که نیازی به اثر مستقیم پروتئین پریون بر پروتئین طبیعی ندارد.
پروتئینی که مغز را نابود میکند
پروتئین غیرطبیعی (پریون) نسبت به عمل آنزیم پروتئاز سلولی (که پروتئینهای اضافی را تجزیه میکند) مقاوم است، بنابراین در مجموع تجمع پریونها در سلول های عصبی رخ میدهد و پس از پلیمریزه شدن این ذرات، فیبریلها (رشتههایی) تشکیل میشود که نهایتاً سلولهای عصبی را تخریب میکنند و برحسب ناحیه ای از مغز که تخریب میشود علائم مربوط بروز میکند.
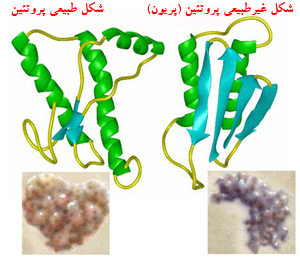
نکته مهم این است که پریون ها باعث بروز واکنش التهابی از طرف دستگاه ایمنی نمیشوند چرا که پریون ها از لحاظ ترکیب شیمیایی مشابه پروتئینی طبیعی در بدن هستند و تنها شکل فضایی متفاوتی دارند، بنابراین "خودی" محسوب میشوند.
ظاهراً برای انتقال پریون ها بین جانوران تماس مستقیم با بافت های مبتلا لازم است. مثلاً در موارد "بیماری کروتز فلد- جاکوب" در گذشته ناشی از انتقال عامل بیماری از راه تزریق هورمون رشد (GH) - که در آن زمان از غده هیپوفیز انسانی استخراج میشد - پیوند قرنیه و نیز از طریق وسائل جراحی مغز بوده است.
پریون ها چون فاقد اسید نوکلئیک هستند، در برابر روشی از استریل کردن در اتوکلاو که در گذشته برای اغلب وسائل جراحی به کار میرفت مقاوم هستند.
امروزه با اجرای دستورالعمل های جدید استریلیزاسیون امکان این انتقال برطرف شده است.
همچنین با توجه به همه گیری جنون گاوی و بروز نوع جدید CJD در جوانان در انگلیس و چند کشور دیگر تصور میرود که مصرف غذایی بافت های آلوده حیوانات (بر حسب آنکه چقدر حاوی بافت های عصبی باشد) باعث تجمع تدریجی پریون ها در بدن در بروز بیماری شود، همانطور که مراسم آدمخواری در انتقال بیماری "کورو" نقش داشت.





















